
Vpr Reprograms T Cells: How HIV-1 Exploits NFAT to Boost Replication
The accessory protein Vpr of Human immunodeficiency virus 1 has long been known to manipulate host cells, but how it reshapes cellular gene expression has remained incompletely understood. In their new publication, the Schindler lab demonstrates that Vpr induces a broad, NFAT-driven transcriptional program in primary CD4⁺ T cells, the main target cells of HIV-1.
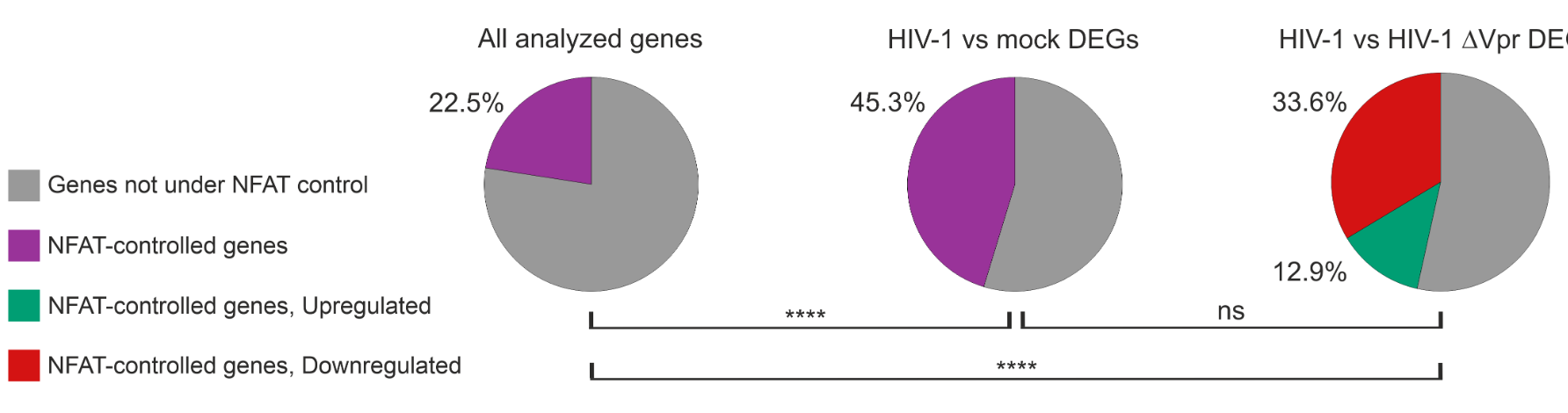
Using RNA-sequencing of infected primary T cells, the researchers compared viruses carrying functional or defective Vpr. Remarkably, 46.5% of more than 1,000 Vpr-deregulated genes were found to be under the control of NFAT (nuclear factor of activated T cells), a central regulator of T cell activation. Vpr stimulated NFAT activity across different HIV-1 groups and subtypes, highlighting the evolutionary conservation of this mechanism.
Functionally, Vpr enhanced the expression of genes linked to immune activation, signaling, and proliferation, while repressing pathways involved in cell cycle progression and chromosome organization. Importantly, pharmacological or genetic inhibition of NFAT alleviated the Vpr‑induced G2 arrest in Jurkat T cells and reduced HIV‑1 replication in primary CD4⁺ T cells.
The study further clarifies the long-debated role of Vpr in cell cycle control: while a clear G2 arrest was observed in immortalized Jurkat T cells, this phenotype was not detected in primary CD4⁺ T cells.
Together, these findings identify NFAT activation as a key regulatory event by which HIV-1 Vpr reprograms its host cell to create an environment favorable for viral replication.
The article was published in mBio and can be found here.
Image: Pie charts illustrating the percentage of NFAT-controlled genes among different gene sets, including genes that are differentially expressed (DEGs) between cells infected with wild type HIV-1 and cells infected with vpr-deficient HIV-1 (pie chart on the right). See Fig. 3 of the publication for details. Cropped from Fig. 3 of © 2026 Leyens et al. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International license.