
Die AG Schürch erforscht die Immunüberwachung von soliden Tumoren und hämatologischen Neoplasien. Ein Schwerpunkt liegt dabei auf der Charakterisierung des sogenannten Immun-Tumor-Mikromilieus (iTME) mittels Hochmultiplex-Mikroskopie und maschinellen Lernverfahren, sowie Bioreaktor-Modellen und Mausmodellen. Unsere Arbeiten sollen dazu beitragen, die Vorhersage eines Ansprechens auf Immuntherapien zu verbessern und immunologische Mechanismen der Tumorkontrolle aufzuzeigen. Zudem beschäftigen wir uns mit der Weiterentwicklung der Hochmultiplex-Mikroskopie und von computergestützten Verfahren für die Datenanalyse.
Advanced Tissue Imaging & Digital Pathology
Kontakt
Leiter
frontend.sr-only_#{element.icon}: Univ.-Prof. Dr. med. Christian Schürch, MD, PhD
Mehr zur Person
Techniken
- Hochmultiplex-Fluoreszenzmikroskopie (50-80 gleichzeitige Marker an einem Schnittpräparat) mittels CODEX (CO-Detection by indEXing)
- Hochpräzisions-Tissue Microarrays (3D Histech GTMA Grand Master)
- Live 3D Gewebe-Bioreaktoren (Cellec Biotek)
- Einzelzell-RNA-Sequenzierung
- Next-Generation-Sequencing
- Leukämie-Mausmodelle
- Maschinelles Lernen und computergestützte Datenanalyseverfahren
- Digitale Pathologie
Projekte
Hochmultiplex-Mikroskopie des Knochenmark-Mikromilieus während der Leukämieprogression und Immuntherapie
Das Knochenmark-Mikromilieu (KMM) ist für die normale Hämatopoese von entscheidender Bedeutung. Bei Leukämien wird das KMM durch leukämische Zellen umgestaltet, die eine dysfunktionale, pro-leukämische Nische hervorrufen, und das Zusammenspiel von leukämischen Zellen mit ihrer Nische ist entscheidend für die Entstehung und das Fortschreiten der Leukämie.
In diesem Projekt untersuchen wir die Leukämieprogression im KM in einem BCR::ABL-induzierten CML Mausmodell. Das KM wird mittels CODEX mit einem Panel von 54 Antikörpern untersucht, um verschiedene Zelltypen, Funktionszustände und zelluläre Nachbarschaften im BMME zu identifizieren. Weiters werden wir Mäuse mit Immuntherapien (anti-PD-1, anti-CTLA-4) behandeln, um die räumliche Zusammensetzung und die Interaktionen der T-Lymphozyten unter Immuntherapie zu verstehen.
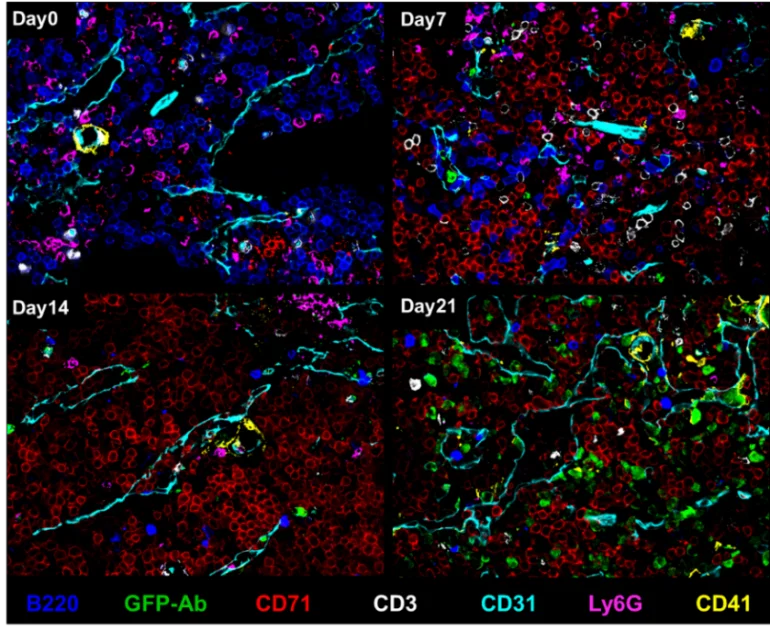
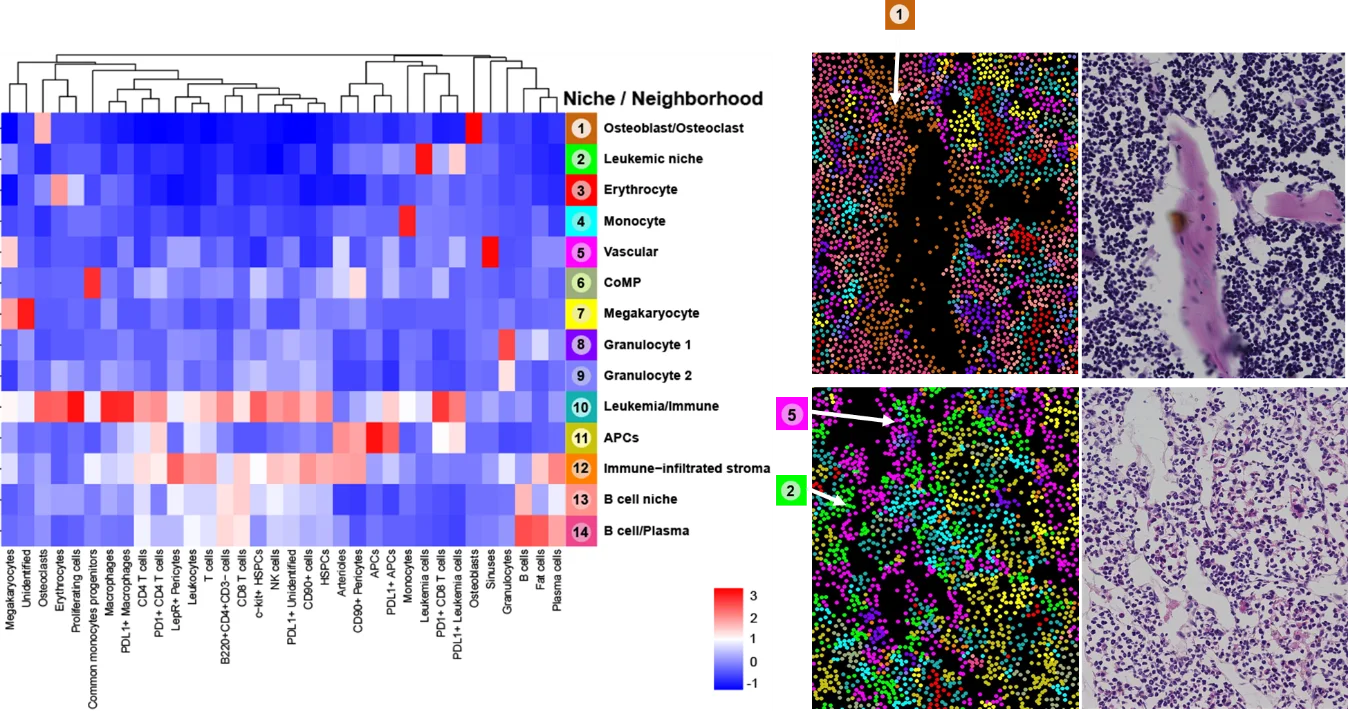
Unsere vorläufigen CODEX-Daten aus Mäusen mit chronischer myeloischer Leukämie (CML) und Wildtyp-Mäusen zeigen eine massive Zunahme vaskulärer Zelltypen, während B-Zellen im Verlauf der Leukämieprogression deutlich reduziert werden (Abb. 1). Unüberwachtes Clustering und anschließende überwachte Zusammenführung von Clustern ergaben 34 Zelltyp-Cluster (Abb. 2). Wir fanden 14 verschiedene Nischen im KMM, die anhand der ursprünglichen H&E-gefärbten Schnitte und Fluoreszenzbilder validiert wurden (Abb. 2). Darüber hinaus ist PD-L1 bei fortgeschrittener CML auf Tumor- und Nicht-Tumor-BM-Zelltypen stark hochreguliert.
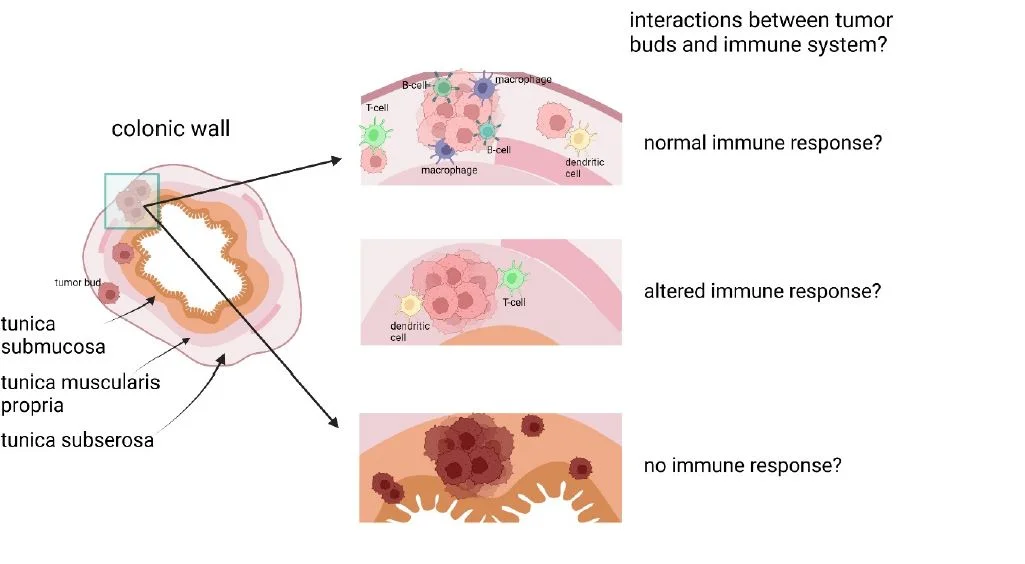
Die Rolle des Gewebe-Mikromilieus bei der Immunreaktion gegen die Tumorknospen („Tumor Buds“) des kolorektalen Karzinoms
Hintergrund: Das kolorektale Karzinom (CRC) ist eine häufige und tödliche Form von Krebs. Die Standardbehandlung umfasst häufig eine Kombination aus biologischen und zytotoxischen Wirkstoffen wie Irinotecan und Cetuximab. Tumorknospen, kleine Cluster oder einzelne Zellen, die sich vom Haupttumor ablösen, haben sich als robuster und signifikanter prognostischer Marker für Darmkrebs erwiesen, da eine hohe Anzahl von Tumorknospen mit einer erhöhten Häufigkeit von Lymphknotenmetastasen oder Fernmetastasen in Verbindung gebracht wird. Tumorknospen treten in erster Linie an der invasiven Tumorfront auf, können aber auch innerhalb des Tumors vorkommen.
Das Verständnis der extrinsischen und intrinsischen Mechanismen der Tumorknospung und der Interaktionen in den verschiedenen Schichten der Dickdarmwand ist noch nicht vollständig geklärt. Um zu untersuchen, wie extrinsische und intrinsische Faktoren die Signalübertragung und den Zyklus von Krebszellen beeinflussen und welche Rolle der infiltrierte Gewebetyp für die Interaktionen in der Tumormikroumgebung (TME) spielt, werden fortschrittliche Bildgebungsverfahren wie CODEX und genetische Analysemethoden eingesetzt.
Dieses Projekt untersucht Tumorknospen, ihre Interaktionen mit dem Immunsystem und dem entsprechenden TME sowie die Rolle des infiltrierten Gewebetyps. Mehr als 50 Biomarker werden gleichzeitig zur systematischen Analyse des Tumorknospungsprozesses in der Dickdarmwand eingesetzt (Abb. 3). Mit dieser Forschung wird es möglich sein, eine wichtige Grundlage für die Entwicklung einer zukünftigen Anti-Budding-Therapie und eines neuen Ansatzes für die antitumorale Immunität zu schaffen.
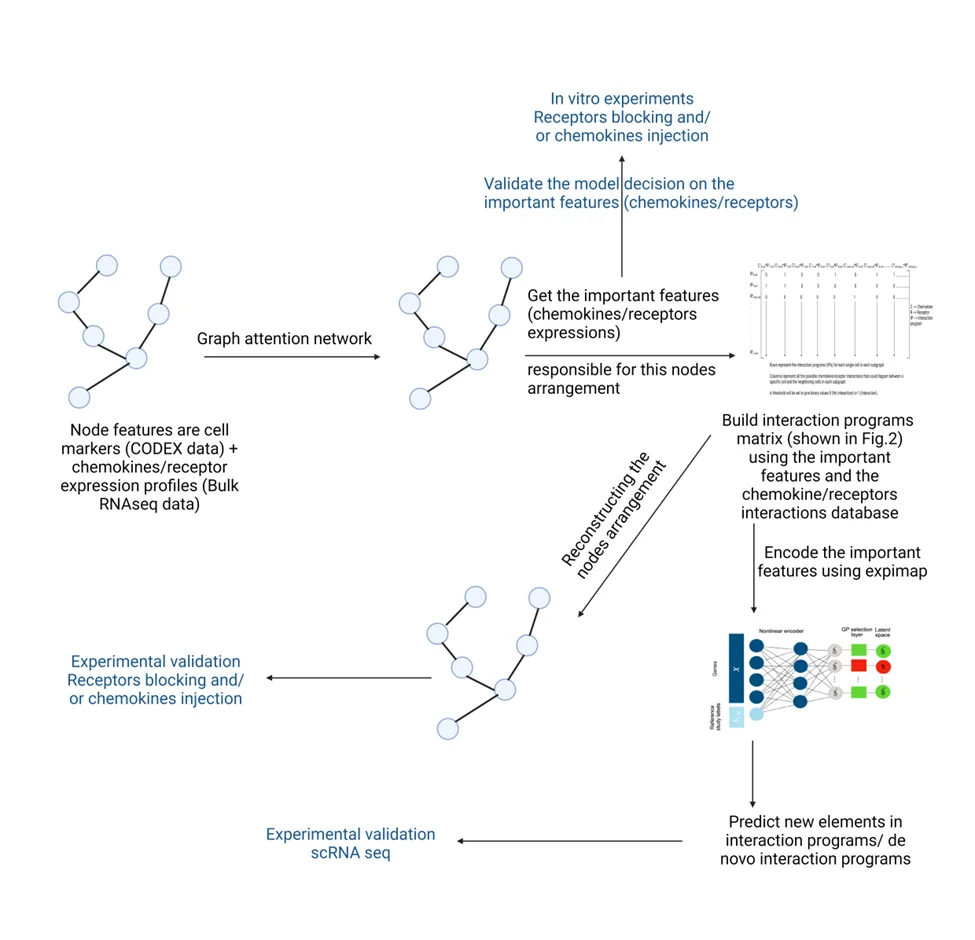
Graph-Neuronale Netzwerke zur Modellierung der Zell-Zell-Kommunikation durch Chemokine im Tumor-Mikromilieu
Chemokine sind eine große Familie von kleinen Proteinen, die für ihre Funktion bei der Zellwanderung bekannt sind. Daher spielen sie eine wichtige Rolle bei der Entwicklung und Homöostase des Immunsystems. Darüber hinaus sind Chemokine an allen schützenden oder zerstörerischen Immun- und Entzündungsreaktionen beteiligt. Das Tumor-Mikromilieu (TME) besteht aus verschiedenen Zelltypen, darunter auch Immunzellen. Interessanterweise korreliert die räumliche Organisation des TME mit dem Ansprechen des Patienten auf eine Immuntherapie.
Graphen sind eine Art von Datenstruktur, die eine Reihe von Objekten als Knoten und ihre Beziehungen als Kanten darstellt. Neuronale Graphen-Netzwerke wurden eingeführt, um die in Graphen gespeicherten Informationen zu erlernen, so dass sie verschiedene Vorhersageaufgaben erfüllen können. Graph-neuronale Netzwerke haben sich bei der Verarbeitung biologischer Bilder als Graphen bewährt und ermöglichen die Vorhersage der Reaktion von Patienten auf eine Behandlung, die Modellierung der Zell-Zell-Kommunikation und die Erkennung von Gewebestrukturen. Darüber hinaus sind interpretierbare Deep-Learning-Modelle, wie z. B. Expimap, eine große Hilfe bei der Entschlüsselung verschiedener biologischer Entdeckungen. Unser Ziel ist es, mit Hilfe von Graph-Deep-Learning eine Korrelation zwischen den räumlichen Informationen des TME und seinen Chemokin-/Rezeptor-Interaktionen herzustellen. Dieser Lernansatz sollte es letztlich ermöglichen, die räumliche Organisation des TME anhand der Chemokin-/Rezeptor-Expressionsprofile vorherzusagen (Abb. 4).
Vorhersage von Krebsimmuntherapie-Ergebnissen durch Anwendung maschinellen Lernens auf hochmultiplexen Gewebebilddaten
Krebsimmuntherapien haben sich in der klinischen Praxis als sehr vielversprechend erwiesen, doch ihre Wirksamkeit ist nach wie vor uneinheitlich, und nur eine Minderheit der Patienten und Patientinnen erreicht ein kuratives Ergebnis. Diese Variabilität wird auf das komplexe und dynamische Tumor-Mikromilieu (TME) zurückgeführt, das die Immunreaktionen behindern und zu suboptimalen Ergebnissen bei der Immuntherapie führen kann. Jüngste Forschungsarbeiten haben die zentrale Rolle des TME bei der Beeinflussung der Wirksamkeit der Immuntherapie unterstrichen und seine Bedeutung für die Vermittlung von Interaktionen zwischen Tumor- und Immunzellen und die Gestaltung der antitumoralen Immunität hervorgehoben.
Die Studie verfolgt zwei Hauptziele. Erstens soll das S3-CIMA-Modell für maschinelles Lernen durch die Nutzung von Datensätzen aus hochmultiplexer Gewebebildgebung mit Schwerpunkt auf Darmkrebs validiert werden. Zweitens versucht die Studie, die Anwendbarkeit des Modells auf die Vorhersage von Immuntherapieergebnissen bei Adenokarzinomen des gastroösophagealen Übergangs (GEJ) auszuweiten.
Die Forschungsmethodik umfasst den Einsatz der CODEX-Technik zur hochmultiplexen Gewebebildgebung, gefolgt von einer sorgfältigen Bildverarbeitung und rigorosen Analyse, einschließlich der genauen Identifizierung verschiedener Zelltypen und der Charakterisierung ihrer räumlichen Beziehungen innerhalb des TME (Abb. 5). Der Aufbau einer robusten Patientenkohorte, die Patienten mit GEJ-Adenokarzinom umfasst, erfordert die Vorbereitung von Proben, einschließlich einer umfassenden und klinisch relevanten Datenerfassung.
Dieses Forschungsvorhaben ist ein entscheidender Schritt, um unser Verständnis der Immuntherapie zu verbessern, neue therapeutische Ziele zu identifizieren und die Onkologie mit wertvollen klinischen Entscheidungshilfen auszustatten.

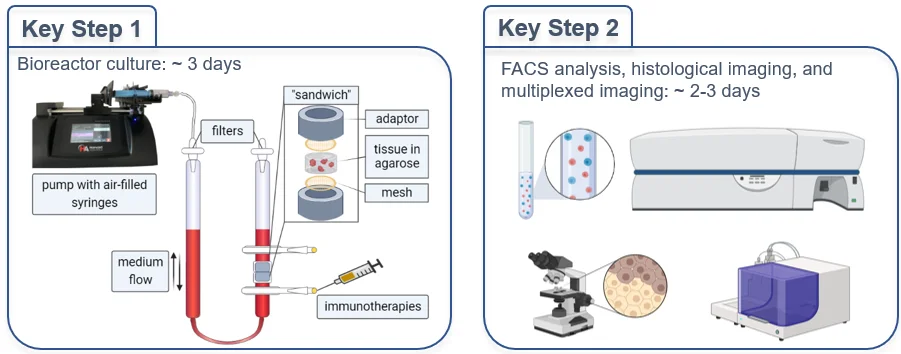
3D-Modellierung der Immuntherapie menschlicher Lymphome in Perfusions-Bioreaktoren mit lebender Gewebekultur
Bösartige Neubildungen wie das diffuse großzellige B-Zell-Lymphom (DLBCL) sind komplexe Systeme, die aus Tumorzellen und dem Tumor-Mikromilieu (TME) bestehen. Die Zusammensetzung und die räumlichen Merkmale des TME haben einen erheblichen Einfluss auf die Ergebnisse der Patienten und Patientinnen, das Ansprechen auf die Therapie und den Krankheitsverlauf. Die Immuntherapie, insbesondere CAR-T und Immun-Checkpoint-Inhibitoren, bietet vielversprechende Möglichkeiten für die Behandlung des DLBCL. Das Ansprechen der Patientinnen und Patienten ist jedoch unterschiedlich, und die Rolle des TME bleibt unklar.
Diese Studie zielt darauf ab, ein Immuntherapie-Modell für menschliches Lymphgewebe mittels 3D in vitro Perfusionskultur zu entwickeln, um zu verstehen, wie die Immuntherapie das TME des DLBCL in vitro beeinflusst (Abb. 6). Wir wollen die Mechanismen des Ansprechens auf die Immuntherapie aufdecken und Responder und Non-Responder identifizieren.
Entschlüsselung des Tumor-Mikromilieus von Blasenkrebs zur Verbesserung des Ansprechens auf eine Immuntherapie
Das Verständnis der Mechanismen, mit denen Krebszellen dem Immunsystem ausweichen, hat zum klinischen Einsatz von Immun-Checkpoint-Inhibitoren (ICI) gegen PD-1 und PD-L1 bei urothelialem Blasenkrebs (UBC) geführt. Trotz der Stratifizierung der Patienten anhand der PD-L1-Bestimmung durch Immunhistochemie sprechen jedoch nur 20 % der mit ICI behandelten Patienten auf die Therapie an. Aufgrund der Komplexität des immunologischen Tumor-Mikromilieus (TME) ist eine gründliche Untersuchung der Interaktionen zwischen Immunzellen und Krebszellen in der TME des UBC dringend erforderlich, um die Auswahl der Patienten für ICIs zu verbessern. Wir untersuchen Gewebe von ICI-behandelten UBC-Patientenkohorten, um Zell-Zell-Interaktionen und die Zellzusammensetzung des TME zu identifizieren, um dessen räumliche Zusammensetzung zu verstehen und um zu ergründen, wie diese mit dem Therapieansprechen zusammenhängt. Wir sind daran interessiert, die verschiedenen Phänotypen der Immunevasion bei UBC zu entschlüsseln. Wir verwenden modernste räumliche Omics-Technologien, einschließlich CODEX, um eine räumlich aufgelöste Einzelzellkarte von UBC-Patienten zu erstellen, die mit einer Immuntherapie behandelt wurden. Die Entschlüsselung der Zelltopographie von behandelten UBC wird fortgeschrittene Immunphänotypen aufdecken, die eine bessere Stratifizierung von UBC-Patienten ermöglichen und das Ansprechen auf die Immuntherapie verbessern werden (Abb. 7).

Tiefgreifende räumlich-funktionelle Charakterisierung von therapieinduzierten Veränderungen in der stromalen und immunologischen Knochenmarksnische bei akuter myeloischer Leukämie
In den letzten Jahrzehnten hat unser Verständnis der Krebsbiologie einen beispiellosen Sprung gemacht. Gleichzeitig sind die Behandlungsergebnisse für viele Patienten mit Leukämie und anderen bösartigen Erkrankungen nach wie vor schlecht. In diesem translationalen Forschungsprojekt werden wir die Mechanismen des Therapieversagens bei akuter myeloischer Leukämie (AML) erforschen, indem wir uns auf die Interaktion von leukämischen Stammzellen (LSCs) mit Stroma- und Immunzellpopulationen in der Mikroumgebung des Knochenmarks konzentrieren.
LSCs sind die Hauptursache für Rückfälle und Therapieversagen bei AML, haben sich jedoch aufgrund ihrer Heterogenität und Resistenz als schwer angreifbar erwiesen. Neben LSC-intrinsischen Faktoren hängt die Fähigkeit der LSCs, nach der Behandlung zu persistieren, von der Induktion einer "leukämischen Nische" im Knochenmark ab. Diese Nische enthält pro-leukämogene Stroma- und Immunzellen, die die LSCs nachweislich vor Chemotherapie und Immunangriffen schützen. Um LSCs wirksam zu eliminieren, müssen wir daher ihre spezifischen Schwachstellen aufdecken. Hier werden wir modernste Methoden einsetzen, um LSCs auf Einzelzellebene zu identifizieren und zu charakterisieren und ihre räumlichen Interaktion mit anderen Zellen in der immunen Mikroumgebung des Tumors sichtbar zu machen (Abb. 8). Dies wird dazu beitragen, neue Marker und molekulare Mechanismen der Behandlungsresistenz zu finden und den Weg für präzisionsmedizinische Ansätze bei AML zu ebnen.

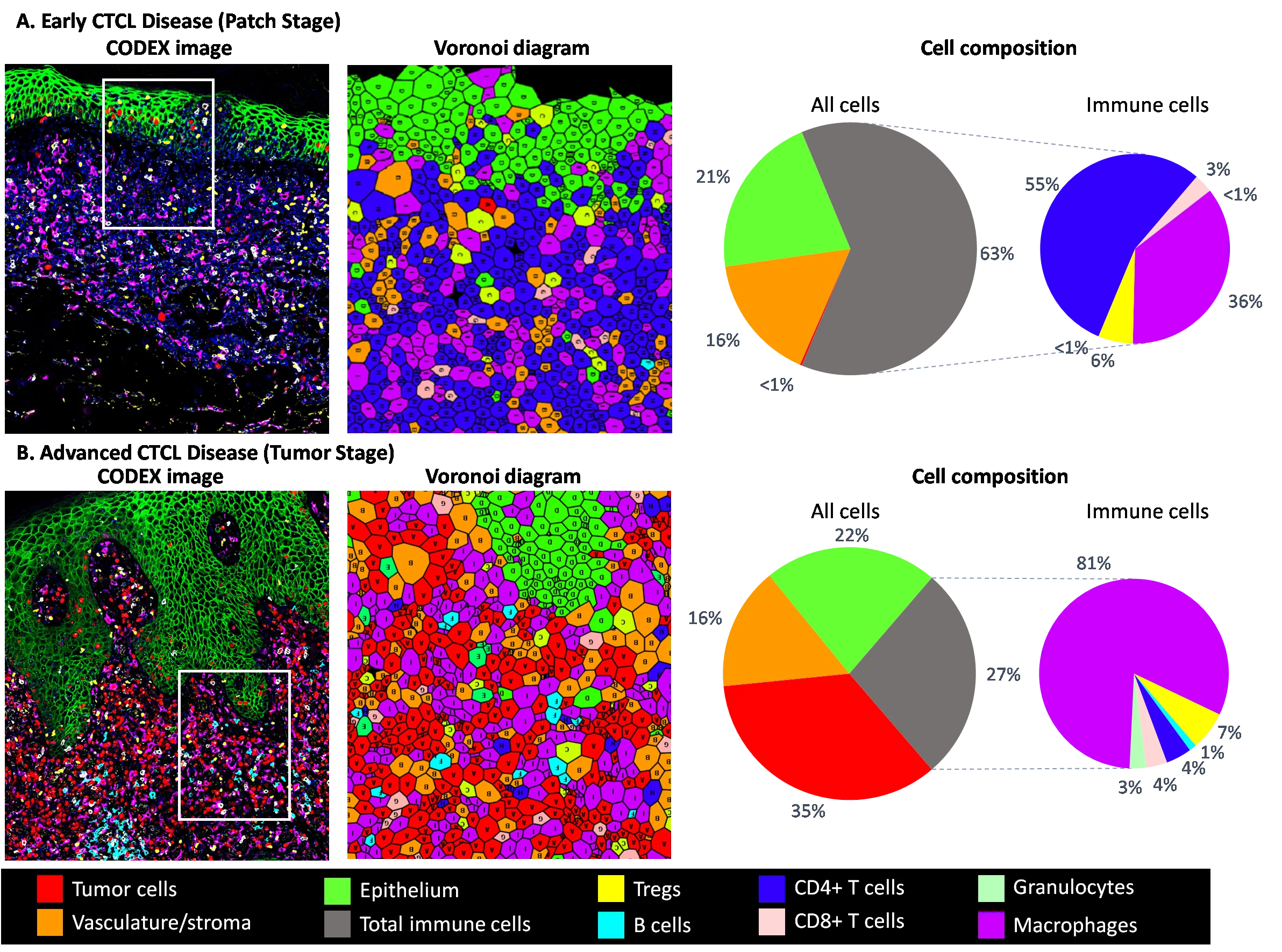
Verbesserte Diagnostik kutaner T-Zell-Lymphome durch hochmultiplex-Mikroskopie
Das kutane T-Zell-Lymphom (CTCL) ist ein seltenes CD4+ T-Zell-Non-Hodgkin-Lymphom der Haut, für das es prinzipiell keine kurativen Therapien gibt und dessen Diagnose im Frühstadium schwierig ist. Eine frühzeitige CTCL-Diagnose ist entscheidend für die rechtzeitige Einleitung einer Behandlung, um ein Fortschreiten der Krankheit zu verhindern.
Das Tumor-Mikromilieu (TME) ist ein entscheidender Faktor für das Fortschreiten und den Ausgang des CTCL. Daher ist eine tiefgreifende Charakterisierung der Architektur von Hautinfiltraten bei frühem CTCL im Vergleich zu entzündlichen Hauterkrankungen auf der Ebene der einzelnen Zelle sehr vielversprechend für die Frühdiagnose, das Verständnis der Mechanismen des Krankheitsverlaufs und die Entdeckung von prädiktiven Biomarkern. Um die Diagnose von frühem CTCL zu verbessern, werden wir in diesem Projekt CODEX Multiplex-Mikroskopie mit transkriptomischer Analyse mittels RNA-Sequenzierung einsetzen, um ein ausführliches Profil von frühen CTCL-Läsionen und gutartigen entzündlichen Hauterkrankungen zu erstellen. Wir erwarten nicht nur, neuartige Kombinationen einiger Marker zu entdecken, die es ermöglichen, die seltenen Tumorzellen in frühen CTCL-Läsionen zu identifizieren, sondern auch spezifische Merkmale zu ermitteln, die den Krankheitsverlauf prognostizieren.
In unseren vorläufigen Daten zeigte die Analyse der verschiedenen CTCL-Krankheitsstadien mittels CODEX nicht nur dramatische Veränderungen im Tumorzellkompartiment, sondern auch in der Zusammensetzung des TME. Während die Häufigkeit von regulatorischen T-Zellen, CD8+ T-Zellen und B-Zellen in Läsionen im frühen und fortgeschrittenen Stadium ähnlich war, war die Häufigkeit von CD4+ T-Zellen stark reduziert zugunsten von erweiterten Makrophagen- und Granulozytenkompartimenten (Abb. 9).
Erweiterung der Multiplex-Kapazität des CODEX-Systems für tiefere Einblicke in Krankheit und Physiologie
Das Erforschen der räumlichen Biologie im Kontext von Erkrankungen und physiologischen Prozessen mittels Hochmultiplex-Mikroskopie bedient sich mehrerer Technologien zur Darstellung von im Gewebe exprimierten Proteinen. Darunter fällt CODEX, in dessen Rahmen Einzelstrang-DNA-gekoppelte Antikörper zur Detektion genutzt werden. Die Darstellung von Proteinen erfolgt durch zyklisches Einführen und Auswaschen von komplementären Fluorophor-gekoppelten Einzelstrang-DNA-Sequenzen.
Besagte Sequenzen sind der limitierende Faktor für die in einem Experiment darstellbare Vielfalt an räumlich exprimierten Markern. Neue Sequenzen werden entwickelt und auf diverse Faktoren wie Kreuz-Reaktivität und Anwendbarkeit im experimentellen Setting ausgetestet (Abb. 10).
Darüber hinaus existieren viele wichtige Marker, die sich mittels „konventioneller“ Färbestrategien nicht ideal im Gewebe darstellen lassen. Diverse Färbestrategien und Konstrukte aus Antikörpern und DNA-Oligonukleotiden werden für die Amplifikation der erfassten Signale im CODEX entwickelt und validiert.

Siehe auch
Zertifikate und Verbände

Focus: Top Nationales Krankenhaus 2026




Stern: Deutschlands Ausgezeichnete Arbeitgeber Pflege


Qualitätspartnerschaft mit der PKV

Erfolgsfaktor Familie

Die Altersvorsorge für den Öffentlichen Dienst
