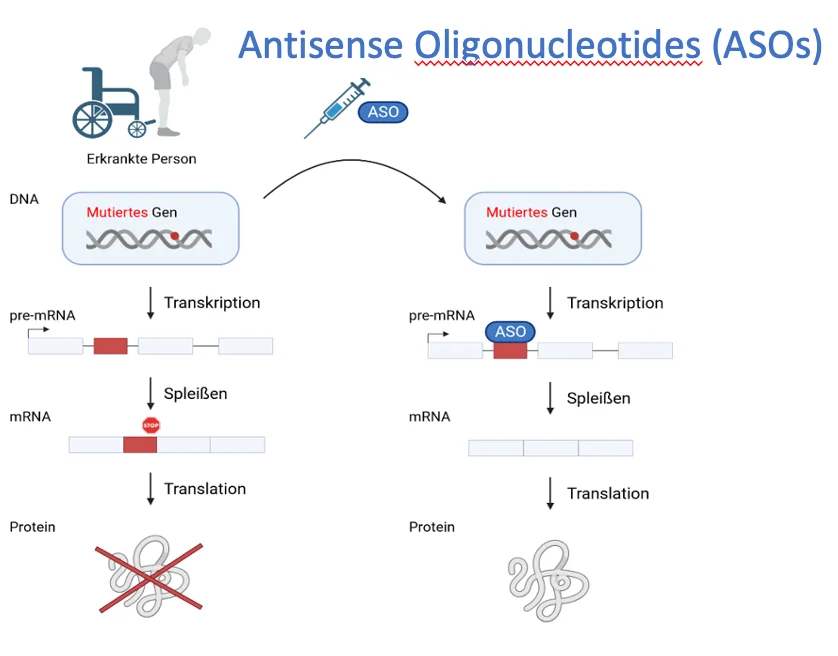
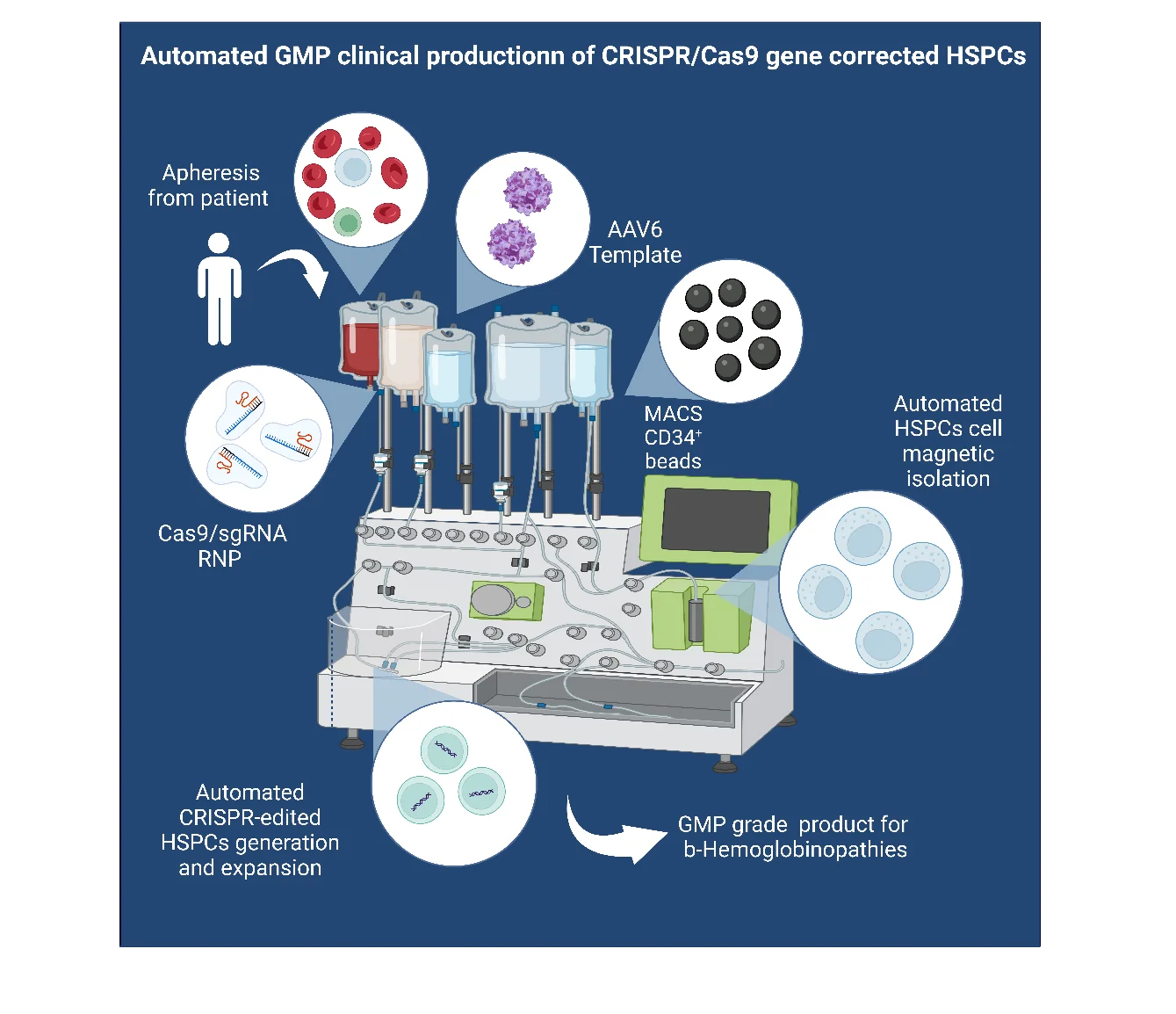
Matthew Porteus (Chair), Professor, Department of Pediatrics- Stem Cell Transplantation, Stanford Medical School, USA.
Annemieke Aartsma-Rus, Professor of Translational Genetics, Department of Human Genetics at Leiden University, The Netherlands.
Alberto Auricchio, Associate Professor of Medical Genetics at the University of Naples and Director of Research at the Telethon Institute of Genetics and Medicine in Naples, Italy.
Christopher Baum, Professor, Chief Translational Research Officer / Chairman of the Board of the Berlin Institute of Health, Charite, Berlin, Germany.
Hildegard Bühning, Professor of Infection Biology and Gene Transfer and Deputy Director of the Institute of Experimental Hematology, Hannover Medical School, Germany. President of the European Society for Gene and Cell Therapy and Scientific Secretary of the German Society for Gene Therapy.
Christine Engeland, Professor of Experimental Virology at Witten/Herdecke University, Germany and Head of the Research Group "Mechanisms of Oncolytic Immunotherapy" at Heidelberg University Hospital, National Center for Tumor Diseases and German Cancer Research Center in Heidelberg.
Boris Fehse, Professor, Head of the Cell and Gene Therapy Research Unit at the Clinic for Stem Cell Transplantation, UKE Hamburg. Scientific Advisory Board (past president) of the Society for Gene Therapy.
Anastasia Khvorova, Professor at the RNA Therapeutic Institute, UMass Medical School, USA.
Martin Meier, PhD, Senior Vice President, Research, Alnylam Pharmaceuticals.
Luigi Naldini, Professor of Cell and Tissue Biology and Gene and Cell Therapy at San Raffaele University School of Medicine. Director of the San Raffaele Telethon Institute for Gene Therapy, Milan, Italy.
Dirk Nettelbeck, PD Dr.,Head of the research group Oncolytic Adenoviruses in the Clinical Cooperation Unit Virotherapy at the German Cancer Research Center (DKFZ) in Heidelberg.