Progressive Familiäre Intrahepatische Cholestase (PFIC)
Beim der seltenen erblichen Lebererkrankung „Progressive familiäre intrahepatische Cholestase“ ist der Galleabfluss gestört. Gallebestandteile sammeln sich in Leber und Blut an. Im späteren Verlauf zeigt sich ein fibrotischer Umbau der Leber bis hin zur Leberzirrhose. Eine Aussicht auf Heilung haben die Kinder nur durch eine Lebertransplantation oder zukünftig wahrscheinlich auch durch eine Gentherapie.
„Progressive familiäre intrahepatische Cholestase“ ist ein Überbegriff für eine Gruppe von Lebererkrankungen bei denen der Galleabfluss gestört ist. Die Krankheit kommt erst zum Vorschein, wenn ein Kind von beiden Eltern ein verantwortliches, verändertes Gen erbt. Das macht sie so selten: Nur eines von 50.000 bis 100.000 neugeborenen Kindern weltweit trägt die Kombination von zwei defekten Genen, die für die Entstehung einer PFIC verantwortlich sind, in sich.
Bei diesen Patientinnen und Patienten treten die Symptome zumeist im Säuglings- bzw. Kleinkindalter auf und entstehen als Folge des gestörten Galle-Transportes von der Leber in den Darm. Die Galle wird in der Leber gebildet und durch die Gallengänge in Richtung Darm transportiert. Dort dient sie der Fettverdauung und der Aufnahme der Fette, einschließlich lebenswichtiger Fettsäuren und Vitamine, in das Blut. Ist der Gallefluss gestört, staut sich die Galle in der Leber und im Blut an.
In vielen Fällen ist die Folge der Störung ein starker Juckreiz, häufig auch Gelbsucht oder eine Gedeihstörung sowie Symptome, die nicht direkt die Leber betreffen. Um den Transport der Galle von den Hepatozyten (Leberzellen) in die Gallekanalikulus zu gewährleisten, befinden sich in der Membran aktive Transportproteine (BSEP, MDR3, FIC1), die unter Energieverbrauch die Gallebestandteile in die Kanälchen pumpen. Defekte in den Genen dieser drei Proteine manifestieren sich in den unterschiedlichen Formen der PFIC (Typ 1-3). Je nachdem welcher Typ vorliegt, sind die Symptome verschieden und mehr oder weniger stark ausgeprägt: Es kommt zu einer Cholestase, dass heißt, dass sich die Gallensäurenkonzentration im Blut erhöht. Daraus resultiert vermutlich der teils extreme Juckreiz (Pruritus), der generalisiert oder lokal begrenzt auftreten kann. Im späteren Verlauf zeigt sich ein fibrotischer Umbau der Leber bis hin zur Leberzirrhose. Bei schweren Verläufen schreitet die Fibrose progressiv voran und schon im ersten Lebensjahrzehnt kann eine terminale Leberzirrhose entstehen, die eine Transplantation notwendig macht. Eine Aussicht auf Heilung haben die Kinder nur durch eine Lebertransplantation oder zukünftig wahrscheinlich auch durch eine Gentherapie.
Diagnostik und Therapie am Kompetenzzentrum
Anamnestisch fallen die Kinder mit PFIC durch häufiges Kratzen und durch den Ikterus auf. Die Symptome beginnen während des ersten Lebensjahres. Sekundär zeigen sich bei den Patientinnen und Patienten Schlafstörungen sowie Wachstums- und Entwicklungsverzögerungen. Labortechnisch zeigt sich zumeist eine signifikant höhere Gallensäuren-Konzentration im Plasma. Die Untersuchung der Leberfunktionswerte lässt weitere Rückschlüsse auf die vorliegende Erkrankung zu: Fast alle Leberenzymaktivitäten und auch die Bilirubinkonzentration im Blut sind bei PFIC im fortgeschrittenen Stadium deutlich erhöht. Eine Leberbiopsie mit anschließender histopathologischer Untersuchung des Gewebes kann Aufschluss geben über den vorliegenden Typ sowie über die Progression der PFIC. In frühen Stadien können die Gallewege noch völlig unverändert aussehen, während in späteren Stadien fibrotische oder bereits zirrhotische Gewebsveränderungen und Gallengangsproliferate zu erkennen sind.

Für die Behandlung der PFIC ist die Erfahrung im Umgang mit dieser seltenen Lebererkrankung unerlässlich. Durch die vergleichsweise hohe Patientenzahl bündeln wir die Expertise von der Diagnostik über die Therapie bis hin zur Lebertranplantation in unserem interdisziplinären Team.
Profitieren Sie von unserer langjährigen Erfahrung:
Kontakt aufnehmen!Das interdisziplinäre Behandlungskonzept beinhaltet neben medikamentösen Therapien, spezielle Ernährungsstrategien und chirurgische Maßnahmen. Die medikamentöse Behandlung bei PFIC ist abhängig von dem Ausmaß der Beschwerden und dient vor allem zu deren Linderung. Am Verlauf der Erkrankung ändern die Medikamente wenig, lediglich das Fortschreiten der Erkrankung kann durch sie verlangsamt werden. Wenn die genannten Therapien nicht die erhofften Linderungen und bei Kindern eine normale körperliche Entwicklung bewirken, kommt es vor allem bei einem raschen und schweren Verlauf der Erkrankung zu schweren Leberschäden. Im Endstadium der Erkrankung muss deshalb die Leber durch eine Transplantation ersetzt werden.
Unser Konzept bei Verdacht auf Progressive Familiäre Intrahepatische Cholestase
Ein wichtiges Therapieziel ist die Verminderung des mitunter sehr belastenden Juckreizes durch die Verbesserung des Galletransports und –stoffwechsels. Es sollen mehr Gallensäuren aus der Leber in die Gallengänge und dann in den Darm transportiert werden. Dort wird die Wiederaufnahme gehemmt und Gallensäuren vermehrt mit dem Stuhl ausgeschieden. Aktuell werden in Studien neue Medikamente für die Nutzung bei PFIC getestet. Sie gehören der Gruppe der Gallensäure-Wiederaufnahmehemmer (IBAT-Inhibitoren) an.
Wenn das Wachstum und die Entwicklung der Patienten durch den Gallensäuren-Mangel im Darm beeinträchtigt sind, ist auf eine konsequente Ernährungstherapie zu achten. Häufig werden Nahrungssupplemente mit mittelkettigen Fettsäuren verschrieben, da diese im Darm auch ohne die Gallensäuren ins Blut aufgenommen werden können. Auch eine parenterale Gabe von langkettigen essentiellen Fettsäuren kann sinnvoll sein. Zur Verbesserung des Vitamin-Status werden fettlösliche Vitamine als Tabletten, Tropfen oder Injektionen verabreicht. In einigen Fällen kann sich auch eine Sonden- oder PEG-Ernährung als vorteilhaft für Wachstum und Gedeihen erweisen.
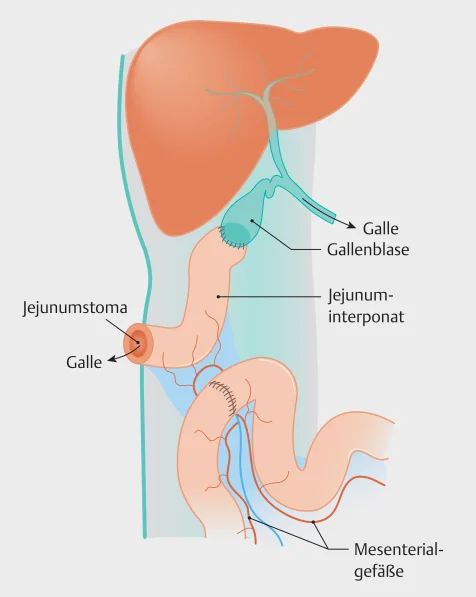
Wenn die Medikamente zur Behandlung des gestörten Galletransportes nicht wirken, können bei geeigneten Patienten die Beschwerden mit Hilfe einer Operation abgemildert werden. Bei der partiellen biliären Ableitung, oder auch Gallengangsdrainage, wird versucht, die Gallensäuren ab- oder umzuleiten, damit weniger Gallensäuren aus dem Dünndarm wieder in das Blut aufgenommen werden. Ein etwa 15 cm langes Stück des unteren Dünndarms wird an die Gallenblase genäht, so wird die Gallenblase mit dem Dickdarm verbunden und das Ende des Dünndarms (Ileum) umgangen. Die Ableitung der Galle erfolgt entweder nach „extern“ durch einen Ausgang (Stoma) zur Bauchdecke (siehe Abbildung) oder „intern“ mit einem Bypass.
Wenn die genannten Therapien nicht die erhofften Linderungen und bei Kindern eine normale körperliche Entwicklung bewirken, kommt es vor allem bei einem raschen und schweren Verlauf der Erkrankung zu schweren Leberschäden. Im Endstadium der Erkrankung muss deshalb die Leber durch eine Transplantation ersetzt werden. Neue Operationsmethoden, wie die Teilung von Spenderorganen oder die Lebendspende eines Teils der Leber durch Verwandte, eröffnen mehr Möglichkeiten für die Lebertransplantation bei Kindern. Sie haben in den meisten Fällen eine gute Prognose und genießen ein hohes Maß an Lebensqualität. Dennoch ist eine lebenslange medikamentöse Behandlung notwendig, damit der Körper die neue Leber nicht abstößt.