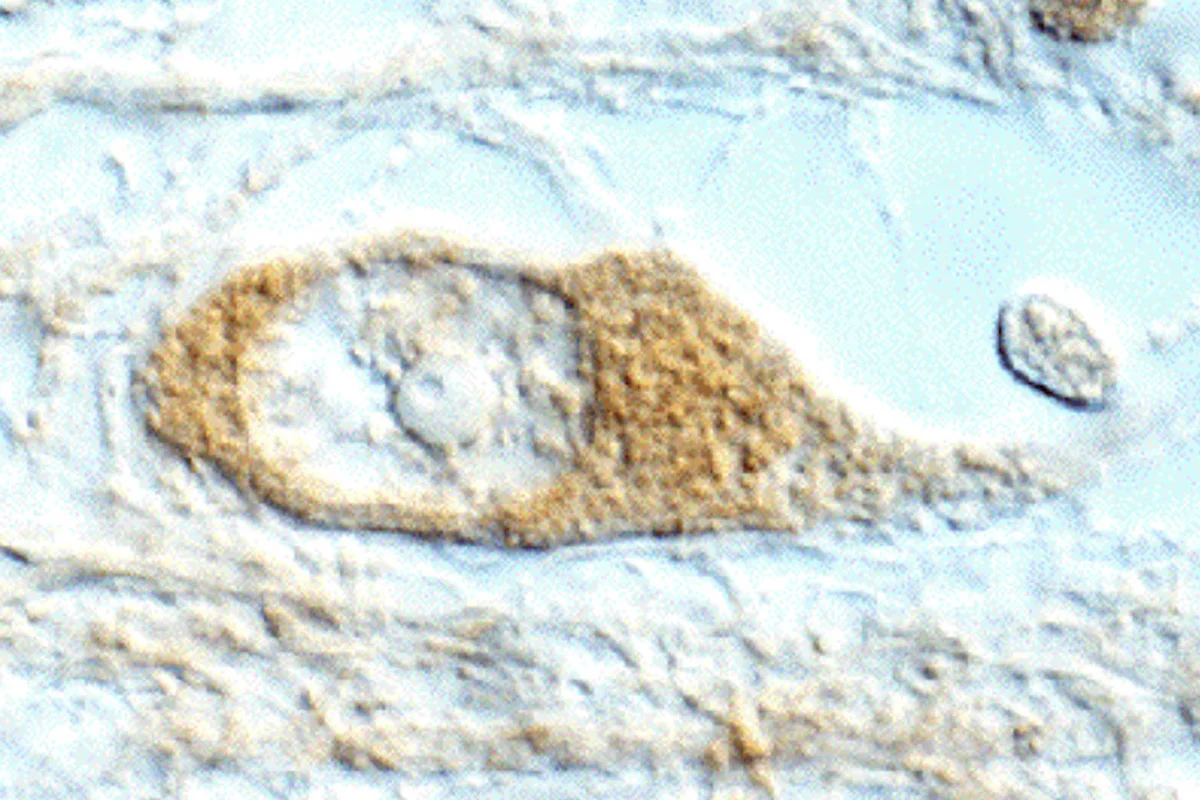
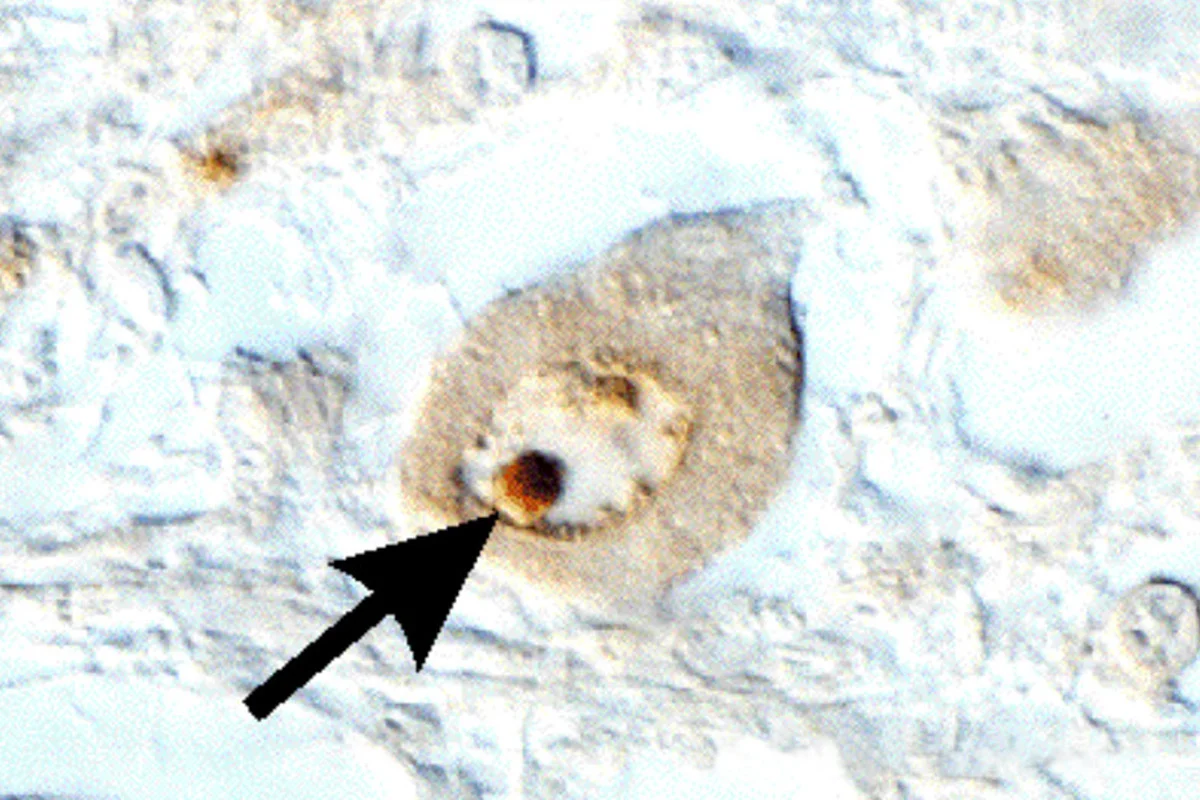
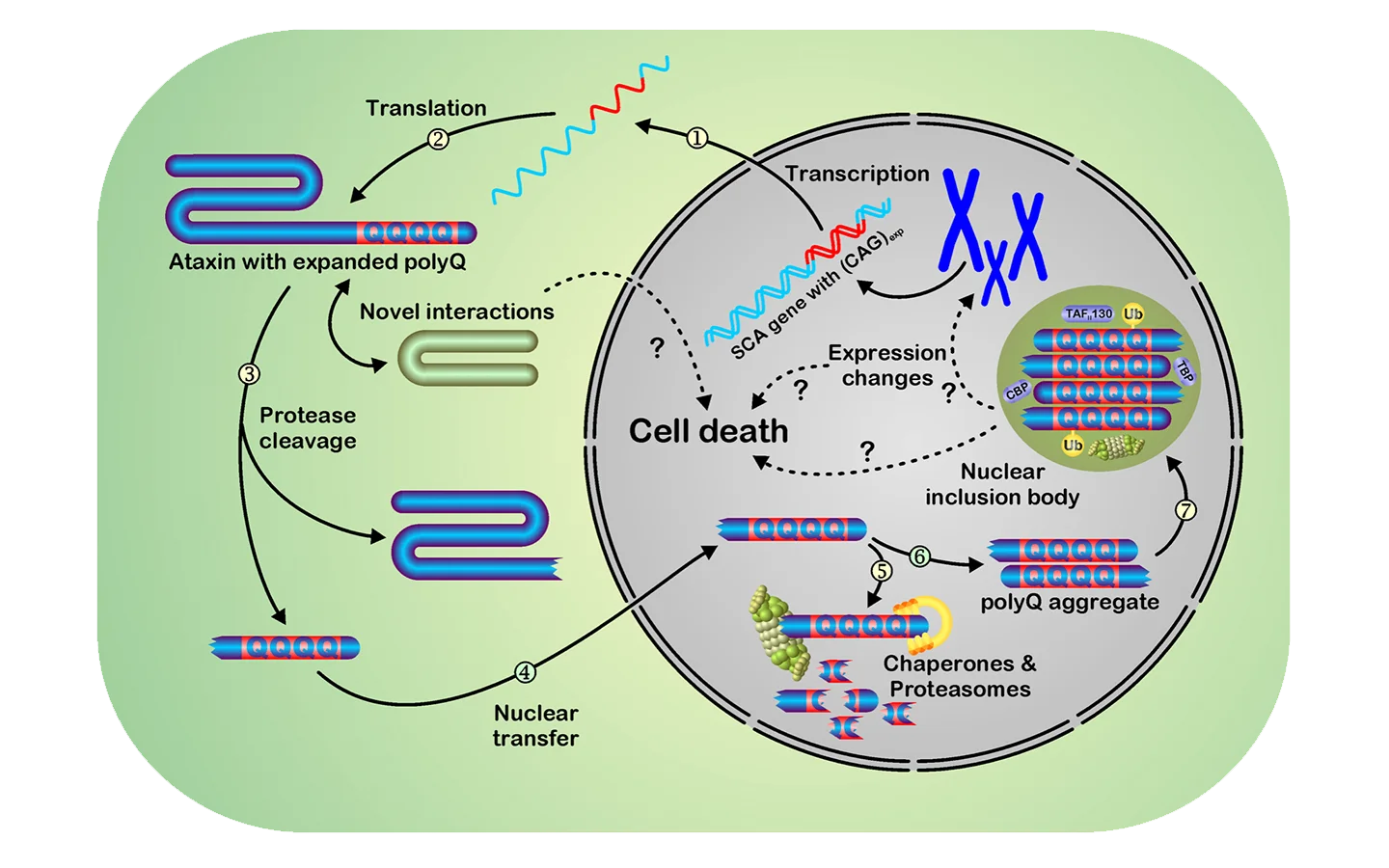
Unsere Arbeitsgruppe beschäftigt sich mit der Spinocerebellaren Ataxie Typ 3 (SCA3), die auch als Machado-Joseph-Erkrankung (MJD) bezeichnet wird. Dabei handelt es sich um eine autosomal-dominant vererbte, spät manifestierende, neurodegenerative Erkrankung, bei der es zu der Bildung typischer sog. intranukleärer Einschlusskörperchen (siehe Abbildung) und einem Absterben von Nervenzellen in bestimmten Regionen des Gehirns kommt.
Ursächlich für die Erkrankung ist die Verlängerung eines Bereiches von CAG-Wiederholungen im ATXN3 Gen. Diese resultiert im kodierten Ataxin-3 Protein in einen expandierten Bereich mit Glutamin-Wiederholungen. Somit gehört die SCA3 zu einer Gruppe von Erkrankungen, den Polyglutamin-Erkrankungen, die alle durch so eine Verlängerung eines Polyglutamin-Bereiches verursacht werden. Weitere Vertreter dieser Erkrankungsgruppe sind u.a. Morbus Huntington sowie weitere Typen Spinocerebellarer Ataxien, wobei es sich bei der SCA3 um die in Deutschland am häufigsten vorkommende Form dieser Ataxien handelt.