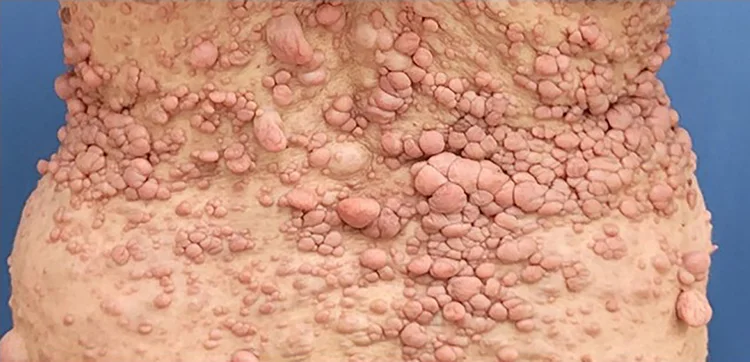
Die Neurofibromatose Typ 1 (NF1, Genveränderung auf dem Chromosom 17) ist im Vergleich zur NF2-bedingten Schwannomatose häufiger anzutreffen und kommt bei ca. einem von 3000 Menschen vor. Das Auftreten ist in ca. 50% erblich und in 50% durch Zufall (Neumutation) bedingt. Im Kindesalter wird die Erkrankung häufig durch das Auftreten von vielen Milchkaffee-Flecken der Haut (Café-au-lait Flecken) in den ersten Lebensjahren diagnostiziert.
Die Diagnose wird (vor allem) klinisch und (häufig ergänzend) genetisch (durch eine Blutentnahme und wenn nötig noch Untersuchung des Tumormaterials) gestellt.
Die Diagnose wird klinisch (durch die Untersuchung) gestellt, wenn mindesten zwei der folgenden Kriterien vorliegen:
|