Die Neurofibromatose Typ 2 (NF 2, Genveränderung auf dem Chromosom 22) ist im Vergleich zur NF 1 viel seltener und tritt bei ca. einem von 40.000 Menschen auf. Die Erkrankung bzw. die Veränderung am NF 2 Gen (Mutation, Verlust der Heterozygosität) ist in ca. 50 % der Fälle ererbt, in den anderen 50% der Fälle spontan neu entstanden.
Im klassischen Fall treten die ersten Beschwerden im Jugend- und jungen Erwachsenenalter auf, jedoch können sich auch schon in der Kindheit „unspezifische“ Beschwerden entwickeln, die häufig nicht mit der NF 2 in Zusammenhang gebracht werden (sog. „Frühsymptome“).
Bei der Erkrankung handelt es sich um ein Tumordispositionssyndrom, bei dem es zur Ausbildung verschiedener Tumore im Bereich des zentralen und peripheren Nervensystems (Meningeome, Schwannome, Ependymome) sowie zu okulären (juveniler Katarakt, retinale Hamartome), ossären (Skoliose, Trichterbrust) und Auffälligkeiten der Haut (kutane Schwannome) kommt.
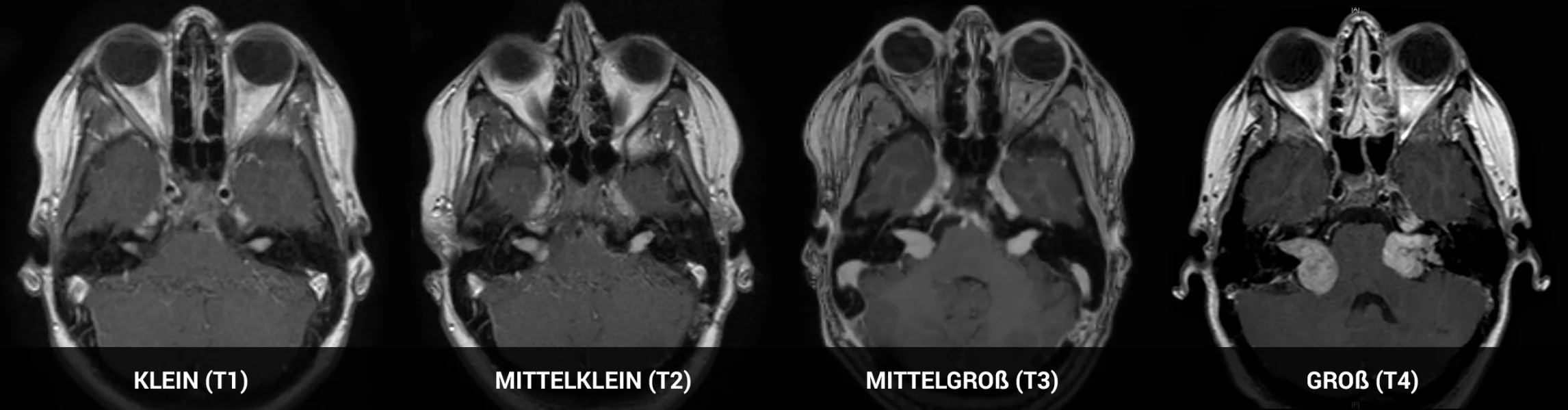
Charakteristisch für die Erkrankung ist das Auftreten beidseitiger Vestibularisschwannome, auch umgangssprachlich als Akustikusneurinome bekannt. Wurde die Erkrankung nicht aufgrund anderer Frühsymptome erkannt, treten die ersten Beschwerden meist durch die Vestibularisschwannome auf, z. B. durch Hörminderung, Hörstürze, Tinnitus, Gleichgewichtsstörungen, Schwindel oder sehr selten Gesichtslähmung (Fazialisparese).