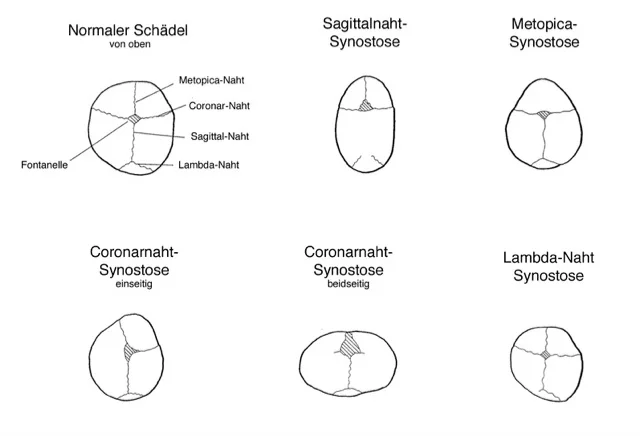
Folgen von Synostosen, Schädelfehlformen und möglichen anderen Fehlbildungen, die in Zusammenhang mit einem Syndrom auftreten können sind:
- Ästhetisch problematische Schädel-Deformierungen, die zu sozialen Ausgrenzungen und Stigmatisierung führen können
- Entwicklung von erhöhtem Druck im Kopf - "Hirndruck"
- Eine zu flache Augenhöhle
- Eine Unterentwicklung des Mittelgesichts
- Begleitende Fehlbildungen an Gehirn, Extremitäten und inneren Organen
- Entwicklungsverzögerungen (inkonstant, je nach Krankheitsbild)
Sehr auffällige Schädeldeformationen wirken oft entstellend und können im späteren Leben zu sozialen Ausgrenzungen führen. Auch bei isolierten Nahtsynostosen ist bei weiterem Schädelwachstum mit einer Zunahme der Schädeldeformation zu rechnen, die das betroffene Kind stigmatisieren und damit beeinträchtigen kann. Deshalb ist unter anderem der ästhetische Aspekt insbesondere bei isolierten Nahtsynostosen wichtig.
Bei einer normalen Entwicklung des Gehirns kann die Wachstumshemmung des Schädels durch eine Nahtsynostose zur einer Steigerung des Hirndruckes führen. Das Ausmaß dieser Hirndrucksteigerung hängt vor allem von der Anzahl der betroffenen Nähte und dem Zeitpunkt der Verknöcherung ab.
Aktuelle Studien unserer Klinik haben gezeigt, dass bei einer vorzeitigen Verknöcherung einer einzelnen Naht, insbesondere der Sagittalnaht, auch stärkere Erhöhungen des Hirndruckes bei bis zu 80% der Kinder auftreten. Unklar ist, ob und wenn ja wie stark dadurch die Entwicklung des Gehirns beeinträchtigt wird. Das Auftreten einer schwerwiegenderen Erhöhung des intrakraniellen Druckes („Hirndruck“) mit der Ausbildung von klassischen „Hirndrucksymptomen“ ist bei einer isolierten Nahtsynostose sehr selten, bei einem Verschluss mehrere Nähte häufiger. Es gibt Hinweise in der Literatur, dass Kinder, die früher an einer Sagittalnahtsynostose operiert wurden (vor dem 6. Lebensmonat) nach 10 Jahren im Schnitt eine bessere kognitive Funktion hatte als Kinder, die spät (nach 12 Monaten) operiert wurden.
Schwere Formen von Kraniosynostosen, wie sie im Rahmen von Syndromen vorkommen, können mit einer Unterentwicklung der Augenhöhlen und des Mittelgesichts einhergehen. Eine unterentwickelte und flache Augenhöhle kann zu einem inkompletten Lidschluss bei stark hervorstehenden Augen führen und dadurch die Hornhaut durch Austrocknung und Entzündung gefährden. Eine Unterentwicklung des Mittelgesichts kann zur Einengungen der Luftwege führen, was sich unter anderem bei Nacht in einem Schlafapnoe-Syndrom äußern kann. Weitere Probleme, die mit einem unterentwickeltem Mittelgesicht einhergehen können, sind: Mittelohrentzündungen und Schwerhörigkeit, falsche Kieferrelation mit einem Zurückliegen des Oberkiefers, Platzmangel der Zähne und Beeinträchtigung der Kaufunktion.
Bei Syndromen muss man mit weiteren Fehlbildungen rechnen, so z.B. Entwicklungsstörung des Gehirns, Innenohrschwerhörigkeit, Verschmelzen von Fingern und Zehen (sog. Syndaktylie) und Gaumenspalten,dem Abrutschen des Kleinhirnes aufgrund eines zu kleinem Kopfe in den Rückenmarkskanal (Chiari 1 Formation) oder eine Blutabflußbehinderung aus dem Schädel.